More
QSAR Certificate Workshop Online of 5 days
Master Quantitative Structure–Activity Relationship (QSAR) modeling from fundamentals to advanced 3D QSAR — learning to build, validate, and apply predictive machine learning models that correlate chemical structure with biological activity
4.7


This Event Includes
- High demand video
- Learn from Experts
- Hands-on practical sessions
- Certificate on completion
Registration Fee |
|
In US $ |
$ 91 |
In Indian Rupees |
₹ 8499 |
Understanding the Field
What Is QSAR and Why Is It Critical in Drug Discovery?
QSAR — Quantitative Structure–Activity Relationship — is a computational modeling approach that uses mathematical equations to describe the relationship between a molecule's chemical structure and its biological activity. By encoding molecular features as numerical descriptors and applying statistical or machine learning models, QSAR allows researchers to predict the biological activity of new, untested compounds before ever synthesizing them in a laboratory.
In modern drug discovery, QSAR is a core tool in the computational chemist's and pharmaceutical researcher's toolkit. It dramatically reduces the time and cost of identifying promising drug candidates by enabling virtual screening of thousands of chemical analogues — predicting which compounds are most likely to be active against a target before committing to expensive synthesis and wet-lab testing.
QSAR now incorporates machine learning algorithms — including linear regression, random forests, and neural networks — making it a frontier discipline at the intersection of computational chemistry, cheminformatics, and artificial intelligence. This workshop makes QSAR accessible and practical for researchers at all levels, using real datasets and the most widely adopted tools in the field.
Workshop Dates
11 to 15 July, 2026. Live trainer will take online sessions from 8 PM - 10 PM India time (+5:30 GMT) time on Zoom.
Complete QSAR Workflow
The End-to-End QSAR Modeling Pipeline
This workshop walks you through every step of the complete QSAR pipeline — from drawing chemical structures to predicting biological activity of new drug candidates.
QSAR Made Simple — Full Pipeline: 2D QSAR → 3D QSAR
Every step performed on your own computer with live trainer guidance · ChemSketch · PaDEL · Excel · WEKA
2D QSAR Pipeline — Days 1–4
📊 Excel Database → ✏️ ChemSketch SDF Structures → 🔢 PaDEL Descriptors → 🔧 WEKA File Preparation → ⚙️Data Processing & Filtering → 🤖 ML Model Building → 📈 Model Evaluation → 🔮 Activity Prediction
3D QSAR Pipeline — Day 5
🧊 3D Structure Preparation → 🔢 3D Descriptor Calculation → 🤖 3D QSAR Model Building → 📊 Testing & Evaluation → 🔮 New Analogue Prediction
Program Overview
What This QSAR Workshop Covers
A 5-day intensive live online training program covering the complete QSAR workflow — from database preparation and descriptor calculation to machine learning model building, validation, and biological activity prediction for new chemical analogues.
⏱️ 90–120 Mins Live Daily- Five days of focused, interactive live sessions — each session building progressively from QSAR fundamentals through to 3D QSAR model building and new analogue prediction.
💻 Work on Your Own Computer- Every analysis step is performed on your own Windows, Mac, or Linux system. All software is free or freely available — the trainer guides installation from Day 1.
🤖 Machine Learning for Drug Discovery- Learn two core machine learning approaches — linear regression and random forest — as applied to QSAR, giving you a foundation in AI-driven computational drug design.
🧊 2D & 3D QSAR Both Covered- The workshop covers both 2D QSAR (Days 1–4) and 3D QSAR (Day 5) — delivering a complete picture of QSAR modeling across both dimensionalities.
🎥 Session Recordings Included- All 5 sessions are recorded and shared after class via YouTube for self-paced revision — revisit any step of the QSAR pipeline anytime.
🏅 Certificate on Completion- Earn a verified BDG Lifesciences certificate with a unique barcode — awarded only upon successfully completing all tasks assigned by the trainer.
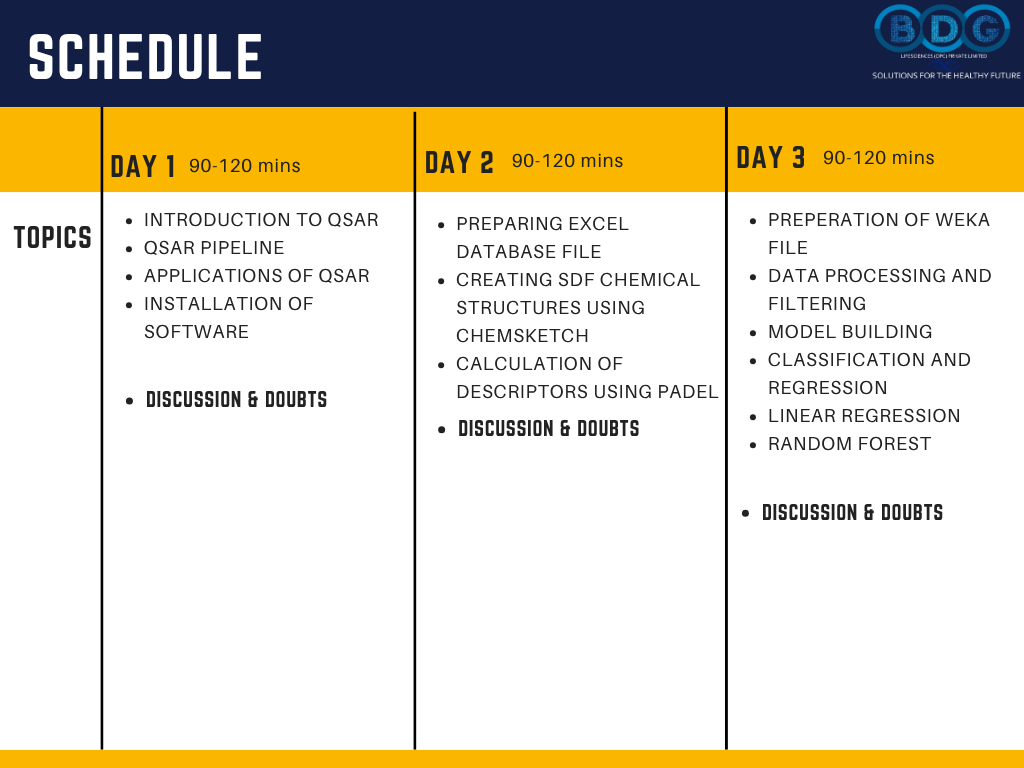
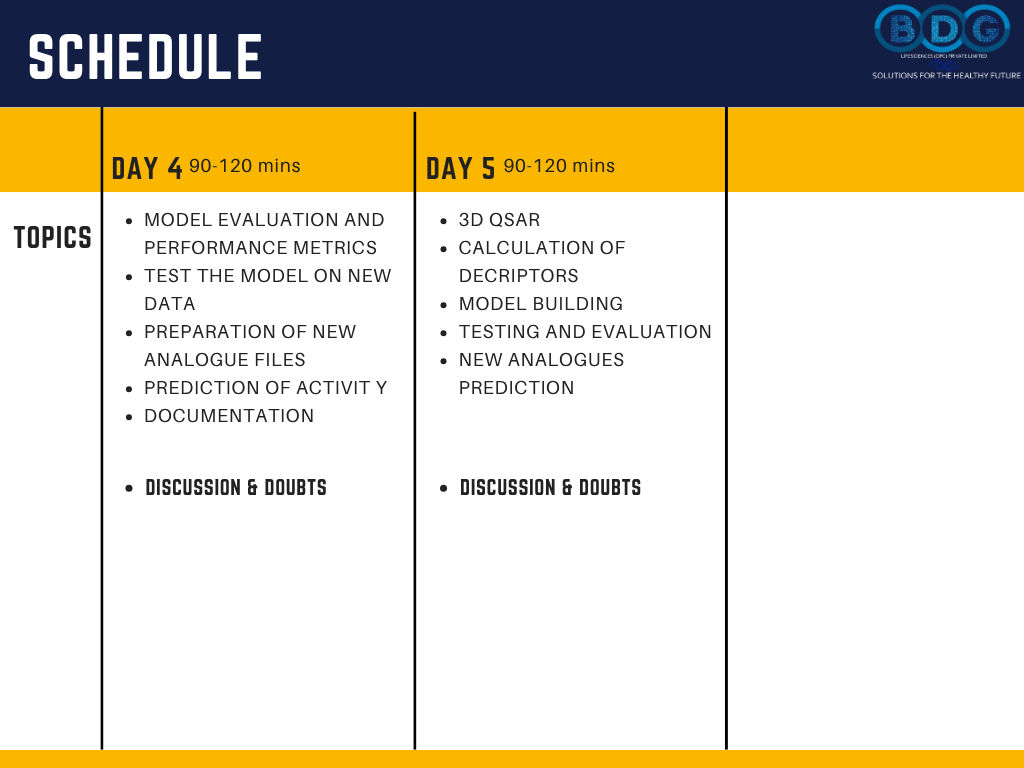
5-Day Schedule
Day-by-Day Workshop Schedule
Five structured days of 90–120 minute live sessions — building progressively from QSAR fundamentals and database preparation through to complete 3D QSAR modeling and new analogue activity prediction. Check out the image section for the daily schedule.
Software & Tools
QSAR Tools You Will Master in This Workshop
Four industry-standard tools — all free — that form the complete QSAR modeling toolkit used in pharmaceutical research, computational chemistry, and academic drug discovery globally.
✏️ ChemSketch (ACD/ChemSketch)- The industry-standard tool for drawing and editing 2D and 3D chemical structures — exporting SDF structure files for descriptor calculation. You will draw and prepare all compound structures in ChemSketch throughout the workshop. Free Software
🔢 PaDEL-Descriptor- The most widely used open-source descriptor calculation software — computing over 1,400 molecular descriptors and 12 types of fingerprints from chemical structure files. PaDEL descriptors form the core input data for all QSAR models built in this workshop. Free · Open Source
🤖 WEKA (Waikato Environment for Knowledge Analysis)- The leading open-source machine learning platform used for QSAR model building in this workshop — implementing linear regression, random forest, cross-validation, and model evaluation workflows through a user-friendly GUI. Free · Open Source
📊 Microsoft Excel / LibreOffice Calc- Used for compound activity database preparation, descriptor data organization, data processing, filtering, and result documentation. Excel is the data management backbone of the QSAR workflow in this workshop. Data Management
All tools are free to download and use. No paid software licenses are required for this workshop. The trainer provides step-by-step installation guidance at the start of Day 1 — arrive without any pre-installed software if preferred.
Why Machine Learning in QSAR Matters for Your Career
QSAR has evolved from classical statistical methods to powerful machine learning approaches — and the pharmaceutical industry, biotech companies, and regulatory agencies are all now requiring computational chemists and drug discovery researchers to have proficiency in both.
This workshop teaches you two of the most important machine learning models used in QSAR — linear regression for interpretable models and random forest for high-performance ensemble prediction — giving you directly applicable, industry-ready skills in AI-driven drug discovery.
QSAR machine learning skills are increasingly required in drug discovery roles at pharma companies, biotech startups, academic research labs, and regulatory bodies such as the FDA and EMA.
Machine Learning Models Covered in This Workshop
- Linear Regression (QSAR) A classical interpretable QSAR model — provides explicit mathematical equations relating molecular descriptors to biological activity, essential for mechanistic understanding and regulatory submissions.
- Random Forest (Ensemble QSAR) A powerful ensemble machine learning model — builds hundreds of decision trees to produce robust, high-accuracy QSAR predictions with built-in feature importance ranking, ideal for complex structure-activity landscapes.
- Classification & Regression Approaches Understand when to use classification QSAR (active/inactive) vs. regression QSAR (continuous activity values) — choosing the right model type for your specific drug discovery objective.
What You Will Achieve
Learning Outcomes After This QSAR Workshop
By Day 5, you will have independently built, validated, and applied both 2D and 3D QSAR models — acquiring the full computational drug discovery skill set needed for research, publications, and industry roles.
- Draw Chemical Structures & Prepare Databases- Use ChemSketch to draw compound structures, export SDF files, and prepare compound activity databases — the essential starting point of any QSAR project.
- Calculate Molecular Descriptors- Compute comprehensive 2D and 3D molecular descriptors using PaDEL — transforming chemical structures into numerical features for machine learning.
- Build & Apply Machine Learning QSAR Models- Train linear regression and random forest QSAR models in WEKA — from data preparation through model building, cross-validation, and external test set evaluation.
- Predict Biological Activity of New Compounds- Apply validated QSAR models to predict the biological activity of new, untested chemical analogues — the core practical skill of computational drug discovery.
- Build Complete 3D QSAR Models- Execute a full 3D QSAR workflow — 3D descriptor calculation, model building, evaluation, and new analogue prediction using three-dimensional molecular features.
- Earn a Verified QSAR Certificate- Receive a BDG Lifesciences certificate with a unique barcode — a credible, verifiable credential for academic publications, PhD applications, and industry job profiles.
Your Expert Instructor
Meet Sharvari Kulkarni
CTO, BDG Lifesciences · Expert in QSAR, Drug Design, Molecular Docking & MD Simulations
Sharvari Kulkarni is a leading bioinformatics expert and the Chief Technology Officer (CTO) of BDG Lifesciences, specializing in QSAR modeling, computational drug design, molecular docking, molecular dynamics simulations, and network pharmacology. She is the only trainer at BDG Lifesciences delivering specialized hands-on QSAR workshops, making this program uniquely valuable for participants seeking expert-led, practical QSAR training.
With a distinguished academic and research profile including 15+ peer-reviewed research publications and extensive experience in computer-aided drug design (CADD), Sharvari brings both rigorous scientific depth and exceptional pedagogical clarity to every session. She is highly proficient in molecular docking across multiple biomolecular targets, and in MD simulations using Schrödinger's Desmond Maestro and GROMACS on both Windows and Linux platforms.
As a workshop trainer, Sharvari is known for her structured, step-by-step lesson plans, patience with participants at all levels, and her ability to make complex computational concepts immediately understandable and practically applicable. She has trained hundreds of participants globally, consistently achieving over 90% positive feedback across all workshops. She also guides research projects, contributes to academic panels, and represents BDG Lifesciences at international scientific engagements.
🧪 QSAR Modeling 💊 Drug Design (CADD) ⚛️ Molecular Docking 🌊 MD Simulations 🌐 Network Pharmacology 🐧 Windows & Linux
15+ Research Papers
90%+ Positive Feedback
Hundreds Trained Globally
CTO BDG Lifesciences
Who Should Attend
This QSAR Workshop Is Designed For
From pharmacy students to computational chemistry researchers and industry scientists — QSAR is a universally applicable skill in drug discovery. This workshop is designed for all experience levels with zero prior QSAR knowledge required.
🎓 Students & Faculty
- M.Sc. & Ph.D. students in pharmacy, pharmaceutical chemistry
- Biotechnology, biochemistry & bioinformatics students
- Microbiology, genetics & life sciences students
- Biomedical technology & immunology students
- Faculty incorporating computational methods into research
- Plant science & agricultural biotechnology researchers
🏭 Industry Professionals
- Pharmaceutical R&D scientists in drug discovery
- Biotech company researchers in computational chemistry
- Regulatory agency scientists evaluating QSAR models
- Bioinformatics professionals expanding into drug design
- Industry professionals seeking AI drug discovery skills
🏥 Researchers & Scientists
- Academic researchers in drug design & medicinal chemistry
- Computational chemistry & cheminformatics researchers
- Pharmacology & pharmacognosy researchers
- Natural product & traditional medicine researchers
- Researchers seeking to add QSAR to publications
No prior QSAR, machine learning, or programming experience required. This workshop uses GUI-based tools throughout — ChemSketch, PaDEL, WEKA, and Excel — making all 23 topics accessible to complete beginners while delivering research-grade skills to experienced participants.
Career Impact
How This QSAR Workshop Advances Your Career
QSAR is a core skill in computational drug discovery — and machine learning QSAR is increasingly demanded across pharma, biotech, regulatory agencies, and academic research positions worldwide.
- Pharmaceutical R&D & Drug Discovery Roles- QSAR modeling is a fundamental skill in pharmaceutical R&D — directly applicable to lead optimization, hit-to-lead campaigns, toxicity prediction, and ADMET modeling roles in drug discovery teams.
- Computational Chemistry & Cheminformatics- QSAR is a core discipline within computational chemistry and cheminformatics — this certificate directly strengthens applications for roles in computational chemistry groups at pharma and biotech companies.
- Academic Research & Publications- QSAR studies are publishable in high-impact journals in medicinal chemistry, pharmaceutical sciences, and computational biology — this workshop equips you to perform and report QSAR studies to publication standard.
- PhD Programs & Research Projects- QSAR skills significantly strengthen PhD applications in pharmacy, pharmaceutical chemistry, and computational biology — and are directly applicable to research projects from day one of enrollment.
- Regulatory Science & QSAR for Compliance- QSAR models are accepted by regulatory agencies (FDA, EMA, ECHA) for toxicity and activity prediction in regulatory submissions — making QSAR skills uniquely valuable in regulatory science careers.
- AI-Driven Drug Discovery — Emerging Roles- Machine learning QSAR sits at the intersection of artificial intelligence and pharmaceutical research — one of the fastest-growing career areas in the life sciences industry globally.
Why Choose Us
Why Choose BDG Lifesciences for QSAR Training?
Since 2010, BDG Lifesciences has delivered research-quality computational drug discovery training globally — with a consistent 4.7/5 participant rating and 2,000+ participants trained across 40+ countries.
✓ The Only QSAR Workshop Taught by a Specialist Trainer- Sharvari Kulkarni is the only QSAR specialist trainer at BDG Lifesciences — with 15+ research papers and hundreds of globally trained participants in computational drug design.
✓ Both 2D & 3D QSAR in One Program- No other online QSAR workshop delivers both 2D and 3D QSAR modeling in a single 5-day program — from linear regression to random forest to 3D descriptor-based prediction.
✓ 100% Free Software Tools- All tools — ChemSketch, PaDEL, WEKA — are free and open-source. No paid software licenses required. The complete QSAR toolkit at zero additional cost beyond the workshop fee.
✓ Government Registered & Internationally Authorized- MSME-registered under the Govt. of India and authorized to operate in Australia & NZ through BBR Group Pty Ltd. (ACN 608 550 849) — a credible, verified training organization.
✓ Live + Recorded — Maximum Flexibility- Attend live for expert guidance and direct Q&A with Sharvari Kulkarni. Access session recordings via YouTube for revision. No analysis step is ever missed.
✓ Publication-Oriented Training- QSAR documentation and reporting covered to academic publication standard — equipping participants to directly incorporate QSAR results into research papers, theses, and journal submissions.
Registration Fee
Simple, Transparent Pricing
One fee for all participants. No hidden charges. Includes live sessions, session recordings, and completion certificate.
Participants in India
₹ 8499 Indian Rupees — same for all Indian participants | Register Now →
Global (Participants Outside India)
$ 91 US Dollars — same for all international participants | Register Now →
Registration is non-refundable and non-transferable. Please read the full T&C below before registering.
FAQs
Frequently Asked Questions
Everything you need to know before registering for the QSAR Made Simple workshop.
What is QSAR and how is it used in drug discovery? QSAR (Quantitative Structure–Activity Relationship) is a computational method that uses mathematical models to predict the biological activity of chemical compounds based on their molecular structure. In drug discovery, QSAR allows researchers to screen thousands of chemical analogues virtually — predicting which ones are most likely to be active against a biological target before synthesizing them in a lab. This saves enormous time, cost, and resources in the drug development process. QSAR is widely used in pharmaceutical R&D, toxicology, environmental science, and regulatory science.
Do I need prior QSAR or programming experience to attend? No prior QSAR, machine learning, or programming experience is needed. This workshop is titled "QSAR Made Simple" for a reason — it starts from absolute fundamentals and builds step-by-step through all 23 topics. All tools (ChemSketch, PaDEL, WEKA, Excel) use graphical user interfaces — no coding or command-line is involved at any stage. The trainer Sharvari Kulkarni is known for her patient, structured teaching style that accommodates participants at all levels simultaneously.
What is the difference between 2D QSAR and 3D QSAR? 2D QSAR uses two-dimensional molecular descriptors — features derived from the chemical graph of a molecule such as molecular weight, topological indices, and electronic properties — to build predictive models. 3D QSAR uses three-dimensional structural features — steric, electrostatic, and shape-based descriptors derived from the 3D conformation of molecules — for model building. 3D QSAR is more computationally demanding but can capture spatial features of the structure-activity relationship that 2D descriptors miss. This workshop teaches both approaches — giving participants a complete picture of QSAR modeling.
What software do I need and is it free? All software used in this workshop is free: (1) ACD/ChemSketch — free chemical structure drawing tool; (2) PaDEL-Descriptor — free open-source descriptor calculator; (3) WEKA — free open-source machine learning platform; (4) Microsoft Excel or LibreOffice Calc (free alternative to Excel). The trainer provides step-by-step installation guidance at the start of Day 1 — you can arrive without any software pre-installed. No paid software licenses are required at any point in this workshop.
Can I use QSAR results from this workshop in a research publication? Yes. QSAR documentation and reporting are covered to academic publication standard in this workshop (Topic 18 — Documentation). QSAR studies using ChemSketch, PaDEL, and WEKA with linear regression and random forest models are regularly published in journals including the Journal of Medicinal Chemistry, Molecular Pharmaceutics, Journal of Chemical Information and Modeling, Computers in Biology and Medicine, and many others. The skills and workflows taught in this workshop are directly applicable to research publications.
Are sessions recorded and what if I miss a day? Yes. All 5 sessions are recorded and shared after class via YouTube using the Gmail ID you register with. If you miss any session, you can complete all corresponding tasks using the recording at your own pace. The program is designed to be self-paced for revision, so missing a session is never a barrier to completing the full QSAR workflow.
Will I receive a certificate after this workshop? Yes. Upon successfully completing all tasks assigned by Sharvari Kulkarni across the 5 days, you will receive a BDG Lifesciences certificate of completion via email. The certificate carries a unique barcode for independent verification and can be printed, shared on LinkedIn, or included in academic and professional portfolios. The certificate is awarded only on task completion — not merely on attendance.
How does QSAR relate to molecular docking and other BDG workshops? QSAR, molecular docking, and MD simulations are three complementary approaches in computational drug design. Molecular docking predicts how a drug candidate physically binds to its target protein. MD simulations study the dynamics of this binding over time. QSAR predicts which chemical structures are most likely to be biologically active — making it the ideal tool for screening large compound libraries before selecting candidates for docking. Together, these three skills form the complete computational drug discovery toolkit. BDG Lifesciences offers dedicated workshops in each: Molecular Docking (10 days), MD Simulation, Drug Design, and this QSAR workshop.
Registration
How To Register
Secure your spot in 3 simple steps — seats are limited for each batch.
1️⃣ Click Register Now- Click the Register Now button and select number of tickets you want to buy..
2️⃣ Check Your Confirmation Email- After successful payment, check your email for a confirmation with registration details and next steps.
3️⃣ Receive Your Zoom Link- Once registration closes, you'll receive the Zoom meeting link and be added to the workshop WhatsApp group.
📧 Need help? Email us at [email protected] or chat with our AI Assistant George at bdglifesciences.com
T & C
- For this event the Fee is same for all participants which is $ 91 US equivalent to 8499 Indian Rupees.
- Please provide a GMAIL ID for registration as the recorded video session will be provided on YouTube. Kindly provide that email ID by which you use YouTube.
- Video recording of each session will be provided at the end of the session to give the user a unique learning experience.
- Interactive training sessions will be conducted on Google Meet/Zoom so to give users a better learning experience.
- As it is with a LIVE TRAINER hence practical application, i.e., the experiments/tasks to be performed can be done in the best explainable manner.
- In this Online Workshop, there will be a LIVE trainer who will solve queries along with training.
- The program is SELF-PACED. After each session video of that particular session will be shared with you so you can go through as many times as you want and perfect yourself in the topics & tasks.
- If you miss any session then NO PROBLEM you can still perform the tasks by going through the video of that particular session.
- Make sure you register under the right category. If you register under the wrong category then your registration will be invalid and NO REFUND WILL BE MADE IN THIS CASE. This is your mistake and the company is not responsible for it.
- To avoid this do read the description of the ticket before selecting and proceeding with payment.
- You need to upload your professional ID which can be your Student ID Card or Research Institute ID Card or Company ID Card. DO NOT UPLOAD EXPIRED ID OR ANY GOVERNMENT ID LIKE AADHAR, PAN, you have to upload professional ID.
- If you are currently not employed or not studying, you may upload the professional ID from your previous affiliation (degree or company), provided you are registering under the correct category. If you are in India, or will be in India at the time of the workshop or training or research project, your category will be Participants in India, and you can upload any valid ID (including one from your previous affiliation). If, at the time of the workshop or training or research project, you are not in India, your category will be Participants outside India, and you can upload any valid ID.
- After registration, you will join the workshop's WhatsApp group. If you have registered in the "Participants in India" category, you must join using an Indian phone number only.
- The certificate will be issued as per the details which you provide in the registration form while registering before payment.
- Once you register relax we will send you the meeting link after the workshop registration gets over.
- We want to make sure that you learn properly hence the training certificate will be given ONLY on successful completion of all the tasks given by the trainer.
- The certificates of all our Online programs are sent by email(softcopy) which has a unique barcode. You can take a print of that on heavy cardstock or photo paper and get it laminated if required.
- The registration is NON-REFUNDABLE and NON-TRANSFERABLE.
- BDG Lifesciences reserves the right of admission in all our programs.
- If you are removed or your registration is canceled then there will be no answer to that. We have our own reasons for such an act of ours
- If we do not wish to give this workshop to any participant then we will refund their amount.
- You should also read the Terms & Conditions page as well as the FAQs page. For any assistance kindly chat with our AI Assistant George on the website www.bdglifesciences.com.
BDG LifeSciences
BDG LifeSciences is a distinguished bioinformatics company established in 2010 and operates globally. Headquartered in India, the company specializes in facilitating workshops, training programs, novel and innovative research projects, and online courses in bioinformatics and life sciences. BDG LifeSciences is registered under the Ministry of MSME (Micro, Small, and Medium Enterprises), Government of India, with the registration number UDYAM-UP-01-0019151. In January 2024, BDG LifeSciences, India, has authorized BBR Group Pty Ltd., Australia (ACN 608 550 849), to operate its programs in Australia and New Zealand.
With a strong focus on the practical application of technology, BDG LifeSciences provides hands-on training where participants work on their own computers/laptops using specialized software and servers. The company has been a leader in this sector for the last 16 years, successfully educating a diverse range of participants, including students, scientists, faculty members, professors, and corporate executives worldwide.
Programs
Related Workshops
- Bioinformatics Workshop
- Drug Discovery & Design
- Molecular Docking Workshop
- MD Simulations Workshop
- WGS Analysis Workshop
- NGS Healthcare Workshop
- Transcriptomics Workshop
Company
- About Us
- Publications
- Advisors & Collaborators
- Blogs
- Careers
- FAQs
- Terms & Conditions
- Privacy Policy
- Contact Us
© 2026 BDG Lifesciences. All Rights Reserved. | 16+ Years of Bioinformatics Excellence | Trusted by 2,000+ Participants in 40+ Countries Privacy Policy · Terms & Conditions
Recommended Just for You




